Computation with
the KEGG pathway database
Hiroyuki Ogata, Susumu Goto, Wataru Fujibuchi, and Minoru Kanehisa
Institute for Chemical Research, Kyoto University,
Uji, Kyoto 611, Japan
Corresponding author:
Minoru Kanehisa
E-mail: kanehisa@kuicr.kyoto-u.ac.jp
Abstract
We introduce and discuss a new computational approach towards prediction and inference of biological functions from genomic sequences by making use of the pathway data in KEGG. Because of its piecewise nature, the current approach of predicting each gene function based on sequence similarity searches often fails to reconstruct cellular functions with all necessary components. The pathway diagram in KEGG, which may be considered a wiring diagram of molecules in biological systems, can be utilized as a reference for functional reconstruction. KEGG also contains binary relations that represent molecular interactions and relations and that can be utilized for computing and comparing pathways.
Keywords: pathway reconstruction,
pathway comparison, sequence analysis, gene organization in the genome
1. Introduction
Living organisms behave as complex systems which are flexible and adaptive to their surroundings. At the molecular level, organisms consist of intricate networks of molecular reactions, which are often called biochemical pathways. Scientists in the fields of biochemistry, molecular genetics, and molecular biology have made great endeavors to uncover various types of biochemical pathways and to abstract common features in different pathways and in different organisms. The best characterized among them are the metabolic pathways that involve enzymatic reactions of chemical compounds. While computational biologists have long been tackling the problem of modeling, simulating, and predicting the metabolic pathways (Franco & Canela, 1984; Brutlag et al., 1991; Cohen & Bergman, 1994; Hofest_dt & Meineke, 1995; Okamoto et al., 1996), the recent explosion of genomic sequence data poses new problems and raises new interests, which is the subject of this manuscript.
At the time of this writing, the complete genomic sequences of nine species are made publicly available. Genomic sequencing has become essential towards elucidating and understanding complicated molecular systems in a cell. Although systematic biochemical and genetic experiments will be necessary in the following phase of functional genomics, computational approaches are also expected to take a large and complementary part of the functional characterization. During the last decade, the sequence databases, combined with the progress in sequence similarity search methods, have proved extremely useful for functional prediction of a single gene or a single molecule. However, in order to characterize the cellular function that results from a network of interacting molecules, a new 'pathway' database must be developed for comparison and computation at a higher level of the biological system. Basically, there are two major problems in representing and computing biochemical pathways.
First, the vast amount of knowledge on molecular pathways that has been accumulated for different cells and different organisms is underrepresented in databases and dispersed over literature. Even in limited attempts to computerize such knowledge, the pathway data are usually entered in databases primarily for browsing purposes. In contrast, we consider that the pathway knowledge should be computerized for the purposes of computation. It is expected that there will be complete genomes of hundreds of species from the three domains of life, Archaea, Bacteria, and Eucarya (Woese et al., 1990; Koonin, 1997). The in silico reconstruction of metabolic pathways is already an essential tool for functional assignment of predicted genes, for almost no data exist by biochemical experiments (Mushegian & Koonin, 1996; Danchin, 1997).
Second, new bioinformatics technologies need be developed to assist functional prediction from sequence data, especially by incorporating systems views on molecular pathways and molecular assemblies. It should be emphasized that the complete genomes of yeast and several bacteria still contain one third to over one half of ORFs that are left uncharacterized because no significant hits are found with well characterized sequences in the existing databases. The threshold of significant hits is somewhat arbitrary and it is often determined with consideration on the expected ratio of true predictions to false predictions (Brenner et al., 1995; Hubbard, 1996). However, a more desirable bioinformatics approach should mimic and automate the biologists' reasoning steps to effectively decrease the threshold when additional biological information is found to be associated with the sequence similarity.
KEGG (Kyoto Encyclopedia of Genes and Genomes) is our new bioinformatics project initiated in 1995 at Kyoto University (Kanehisa, 1997a). KEGG aims at:
2. Pathway Database
The KEGG pathway database consists of two sections: the metabolic pathway section and the regulatory pathway section. While the current knowledge of metabolic pathways is well organized in KEGG, the organization of regulatory pathways is still rudimentary. The metabolic pathway data were first entered from the book "gMetabolic Maps" compiled by the Japanese Biochemical Society (Nishizuka, 1980, 1997) and the wall chart of "Biochemical Pathways" by Boehringer-Mannheim (Gerhard, 1992). They were then verified and updated by using mostly the EMP/WIT database (Selkov et al., 1996) and the 13 volume Enzyme Handbook (Schomburg and Salzmann, 1990).
At the moment KEGG contains about
100 metabolic pathway diagrams grouped into 10 categories. Each diagram
can be retrieved by the hierarchical text menu, by the hierarchically drawn
graphics menu, or by the key word search using DBGET/LinkDB (Kanehisa,
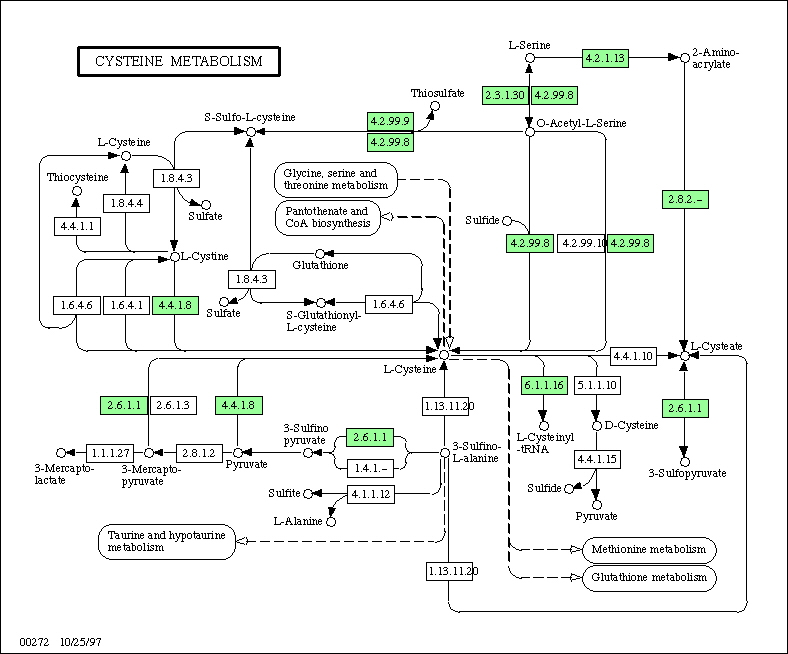
1997b) via the KEGG WWW server (http://www.genome.ad.jp/kegg/). Figure
1 shows an example for cysteine metabolism. There are two characteristics
of the KEGG metabolic pathway diagrams that are relevant to computation.
Figure 1. An example of the KEGG
pathway diagram that represents the relationships of genes or gene products,
in this case, in the pathway of cysteine metabolism. A rectangle is a gene
product (an enzyme) and it is marked (shaded) when the corresponding gene
is found in the genome, in this case, of Escherichia coli.
First, an enzyme denoted by a rectangle is an object that can be manipulated by machine. For each pathway diagram there is one reference diagram which is manually drawn and updated, and all organism specific diagrams are computationally derived by matching the enzyme objects and the corresponding genes in the gene catalog. In this way KEGG is able to immediately reconstruct organism-specific pathways for an increasing number of complete genomes. The enzyme object is hyperlinked to the ENZYME section of the LIGAND database containing, among others, the enzyme nomenclature, the reaction scheme, the chemical compounds involved, and additional links to molecular and biological information (Suyama et al., 1993; Goto et al., manuscript in preparation).
Second, the pathway diagram is drawn
to lay stresses both on the successive conversion of major compounds and
on the appearance of two consecutive enzymes in the pathway. The information
is computerized in the form of, what we call, binary relation, which is
the most fundamental data type in KEGG. The substrate-product binary relations
have been utilized in computing possible reaction paths (Goto et al., 1996)
and the enzyme-enzyme binary relations have been correlated with other
types of molecular relations (Ogata et al., 1996). A similar concept is
the predecessor-list in EcoCyc (Karp et al., 1996) which is utilized for
automatic drawing of metabolic pathways.
3. Pathway Computation
3.1 Reconstructing organism-specific pathways
An organism-specific pathway is automatically
generated by matching the reference pathway diagram and the gene catalog
according to the EC number. When the gene for an enzyme exists in the gene
catalog, the box representing the corresponding enzyme is marked by color
on the pathway. The consecutive appearance of the colored boxes would then
be considered an organism-specific pathway (Fig. 2). In order for this
procedure to be successful, the reference diagram should contain all known
alternatives of reaction paths rather than just the consensus alone. In
fact, we are learning from the complete genomes that different organisms
have different sets of metabolic pathways that reflect the living environment
and the strategy of adaptation. The automatic reconstruction of organism-specific
pathways is a first step toward establishing a well verified set of reference
diagrams.
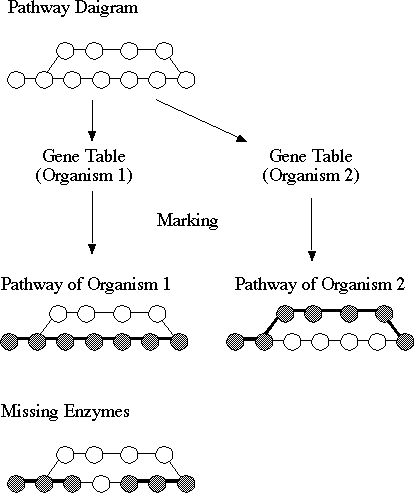
Figure 2. A schematic illustration
of generating organism-specific pathways. The reference pathway diagram
is marked by mapping the information in the gene table on to the components
in the pathway, resulting in a sequence of marked genes that can be considered
an organism-specific pathway. Missing enzymes are the unmarked components
that split the organism specific pathway.
It is often the case that, when all the predicted enzyme genes are mapped on the KEGG reference pathway diagram, the organism-specific pathway would not become continuous or complete because of missing enzymes (Fig. 2). It is possible that the initial gene finding and functional assignment have not been correctly done and missed the enzyme that is actually present in the genome. By changing the threshold values and other parameters the gene may be identified. Alternatively, our knowledge on enzymatic reactions is not sufficient, and an enzyme with a given EC number may function for substrates with wider specificity than previously known. In fact, the number of aminotransferases that appear at the last step of amino acid biosynthesis seemed different in different bacteria and it was necessary to assume that, for example, aspartate aminotransferase catalyze reactions for other amino acids as well (Bono et al., submitted for publication). Moreover, there is also a possibility that our knowledge on metabolic pathways is not sufficient, and alternative paths of reactions may lead to the conversion of the initial substrate to the final product in the missing portion. We formulate this problem in terms of logic programming as described below.
3.2 Computing paths of chemical reactions
In general an enzymatic reaction
involves multiple substrates and multiple products. In KEGG a reaction
is represented by a collection of binary relations between all possible
substrate-product pairs (Goto et al., 1996). Here we consider the simplest
case of one substrate - one product reaction. As formalized below, the
reaction catalyzed by a specific enzyme is a factual data, while the path
is defined in the form of a rule:
reaction( X, Y, E )where X, Y, Z are the chemical compounds, E is the enzyme, and EL is the list of enzymes. The rule says that the path can be composed of just one reaction or it can be derived by extending an existing path by one reaction. As will become apparent in the next subsection 3.3 this formalization is not limited to chemical reactions; namely, successive additions of binary relations can be used to deduce different types of paths.path( X, Y, [E] ) <- reaction( X, Y, E )
path( X, Y, [E | EL] ) <- reaction( X, Z, E ), path( Z, Y, EL )
We have developed a path computation program, PathComp, to efficiently perform linking of binary relations (Goto et al., 1996; Fujibuchi et al., 1997). The implemented algorithm is based on a breadth first search and PathComp provides several options for the calculation. For example, it calculates all paths starting from a given initial substrate, all paths between an initial substrate and a final product, and the shortest paths along a set of compounds. PathComp generates candidates for organism-specific pathways or all chemically feasible pathways either by limiting the reaction data for the enzymes in the gene catalog or by using all the reaction data extracted from the LIGAND database, respectively.
As mentioned, the substrate specificity
of an enzyme in one organism could be different from that of the same enzyme
in another organism. According to the hierarchical classification of EC
numbers, this implies that it may be necessary to include other reactions
in the same category for the enzyme gene predicted from the genome. This
is defined by another rule:
reaction( X, Y, E' ) <- reaction( X, Y, E ), group( G, E, E' )
where enzymes E and E' belong
to the same group G, and this procedure is called query relaxation (Gaasterland
et al., 1992). In PathComp the relaxation can be done according to the
EC number classification, the grouping of sequence motifs, the superfamily
(sequence similarity) classification, and the 3D fold classification.
3.3 Computing paths of reasoning
Various kinds of reasoning steps
during in silico analysis taken by biologists often involve sophisticated
navigation across many links among biological entities. The nature of these
links would be classified into three categories shown in Table 1: factual,
similarity, and biological links (Kanehisa, 1997b). Automation of the reasoning
steps by path computation of these links is one of the major objectives
of the KEGG project. The synthesis of metabolic pathways described above
is a straightforward application of utilizing a well-defined category of
biological links, namely, the substrate-product binary relations.
Table 1. Three type of links
used in path computation.
| Type | Example |
| factual link | cross-references
in databases
links between genes and functions |
| similarity link | sequence
homology (orthology/paralogy)
3D similarity and complementarity |
| biological link | substrate-product
relations in enzymatic reactions
interacting molecules in a cell neighboring genes in the genome |
The similarity search against sequence databases is currently the only automated portion to assist biological reasoning for newly determined sequences. Although the links to other databases may be provided, the biological information associated with the similarity found is strongly dependent on texts that can only be understood by human. KEGG attempts to automate the latter portion of reasoning by computerizing different types of biological links, including molecular counterparts in the pathway or closely associated genes in the genome. Because sequence similarity is just another type of binary relation in KEGG, the entire paths of reasoning can be computed in a similar way as the path computation of chemical reactions.
Figure 3 shows a specific example,
where the reasoning step is defined as a sequence of combining three different
links. Suppose that any functional clue is sought for a newly determined
human gene sequence. By using the similarity links of this gene, homologues
in other organisms can be found. The biological links provided by KEGG
would then identify biological partners in the pathways of these organisms.
By using the similarity links back to human, it is possible to find candidate
genes or gene products that may be interacting with the initial gene or
the gene product in the biological pathways. This and other types of reasoning
steps are currently being implemented by using the DBGET/LinkDB system
(Fujibuchi et al., 1997). However, the most critical aspect is the organization
and computerization of actual data on biological links that are dispersed
over organisms and over literature. The current version of DBGET/LinkDB
(http://www.genome.ad.jp/dbget/dbget.links.html) contains only the enzyme-enzyme
relations in the metabolic pathways.
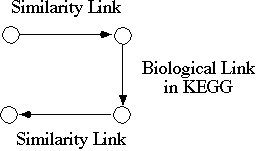
Figure 3. An example of the paths
of reasoning that can be computed with a combination of various kinds of
links.
Chromosomal locations of genes is
also informative for elucidating regulation or evolution of metabolism
in yeast and bacteria (Wolk & Butler, 1997; Otsuka et al., 1996). In
KEGG the location and the order of enzyme genes in the genome can be examined
graphically, as well as the location and the order of enzymes in the pathway.
The example in Figure 4 shows that a set of enzymes coded in close positions
along the E. coli genome forms a block of reaction paths in the
metabolic pathway, which is actually a general tendency as described below.
Although the feature is not provided yet, the positional correlation in
the genome, which is also represented by binary relations, will be included
in the automatic biological reasoning process in KEGG.
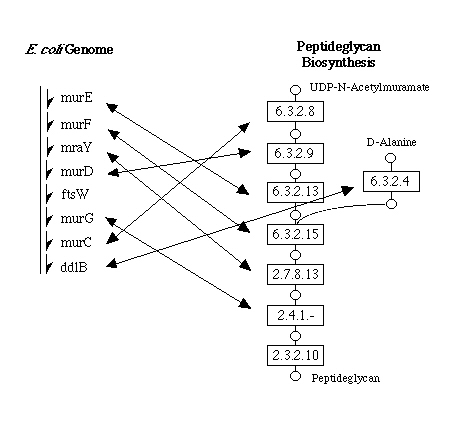
Figure 4. The cluster of genes
in the E. coli genome is often well correlated with the cluster
of enzymes in the metabolic pathway as in this case of peptideglycan biosynthesis.
This type of correlation can be examined graphically in KEGG.
3.4 Comparing biological networks
The previous two subsections 3.2 and 3.3 described apparently the same kind of computation. Here we present another type of computation, which can be categorized as the comparison of biological networks. Here the network includes not only the metabolic and regulatory pathways, but also the genome which is a one-dimensional network of genes and the hierarchical network of evolution formed by orthologous genes. By comparing the same type of networks, for example, the metabolic pathway diagrams, it is possible to identify similarities and variations among different species. By comparing different types of networks, for example, the metabolic pathway and the genome, it is possible to identify groups or clusters of genes that may be considered modules of the biological system.
In bacterial genomes it is known
that functionally related genes tend to be clustered more often than unrelated
genes (Tamames et al., 1997). We searched the enzymes that are coded in
close positions along the genome and that play their roles in close positions
on the metabolic pathways. The functionally related enzymes coding segments
(FRECSs), as we call them, are obtained as shown in Figure 5(a). The path
length between two enzymes is defined by the shortest path calculation
of the PathComp program using the enzyme-enzyme binary relations that represent
the knowledge of actual metabolic pathways in KEGG. The path length is
calculated for each enzyme pair that appears within the sliding window
along the genomic sequence. Figure 5(b) shows the mean path length of the
enzyme pairs within the window sizes of 1 Mbp and 10 kbp for the E.
coli genome that contain, on the average, one gene per 1 kbp. First
of all, the average value for all windows decreased from 5.7 (dotted line)
to 4.5 (solid line) with the decrease in the window size. We considered
that this correlation would be due to the existence of relatively short
(~10 kbp) clusters of related enzyme genes. By marking the related enzyme
gene pairs with the path length of 3 or less and by merging the overlapping
windows of 10 kbp containing related enzyme genes, we obtained the total
of 82 FRECSs. Among them about 54% were found to be related to known operons.
The enzymes in the peptideglycan biosynthesis shown in Figure 4 was one
of the examples.
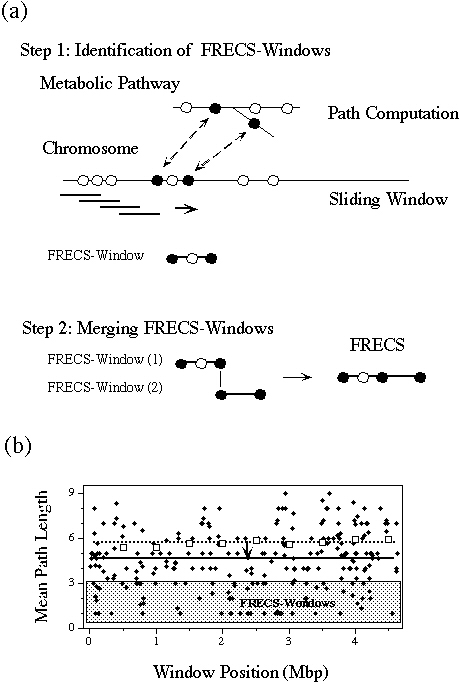
Figure 5. (a) A functionally related
enzymes coding segment (FRECS) is defined by the enzyme pairs that appear
within a sliding window in the genome and that are separated by given path
lengths in the metabolic pathway (see text). (b) The mean path length of
enzyme pairs is plotted against the window position for two different window
sizes: 1 Mbp (white boxes) and 10 kbp (black boxes). The dotted and solid
lines show the average levels of the path length for 1 Mbp and 10 kbp windows,
respectively, and an arrow is put to indicate the difference of the levels.
FRECS corresponds, in this case, to the path length of 3 or less.
Although this analysis focuses on
a comparison of clustering based on two biological links representing pathways
and genomes, a similar comparison can be applied to other types of links
and networks. We observed the correlation of clusters between the biological
links of metabolism and the sequence similarity links; namely, there was
a tendency of paralogous genes appearing closely in the metabolic pathway
(Ogata et al., 1996). There were also cases where apparently similar sets
of enzymes formed similar reaction paths when local similarity patterns
of metabolic pathways were searched (Ogata et al., manuscript in preparation).
We are extending this type of network comparison for analysis of other
problems involving evolution (sequence similarity), pathway formation,
and genome organization.
4. Discussion
In this paper, we have described a computational approach that is intended to assist in silico investigation of gene functions and molecular pathways. This has become feasible because of the unique pathway database being organized by KEGG. A biochemical pathway is an abstraction of a subset of intricate networks in the soup of interacting biomolecules (Mavrovouniotis, 1995), and the abstraction is arbitrarily chosen by biologists at the level of their interests. KEGG*fs pathway is at a highest level of abstraction where only the relation of gene products is emphasized without much describing the detailed molecular processes involved. This is because KEGG's major concern is to cover all aspects of biochemical pathways and to correlate them with the genomic information. This breadth first approach is suitable for analyzing overall behaviors of a biological system, and is complementary to the traditional depth first approach for analyzing specific components of a biological system.
Somewhat similar to the level of abstraction, the experimental data should also be viewed in a hierarchical way. In contrast to the traditional experiments in molecular biology, the experiments such as the systematic analysis by the yeast two-hybrid system (Luban & Goff, 1995) produce, more or less, qualitative data that are accumulated in much larger amounts and at much faster rates. In order to capture an overall picture of the biological system, the progress in data representation and computation of such kinds of data is necessary.
Most of the current computational tools in molecular biology are strongly dependent on the structural information of molecular sequences and 3D coordinate data that are relatively simple to represent. In contrast, it is not an easy task to represent functional information for computation by machine, although it is not difficult to describe function by texts for human to understand. We consider the essence of function lies in the interaction of molecules and adopted the representation of binary relations. The network is an abstraction of molecular pathways, gene organization in the genome, and functional and structural hierarchies of molecules, all of which is considered to result from a set of binary relations. Furthermore, the network is also an abstraction of the chain of logical reasoning steps by human, or the deduction from a set of relations. Thus, the path computation techniques and the network comparison techniques based on binary relations will have a broad range of applications.
The benefit of whole genome sequencing
is the completeness of the ORF catalog, even though it may contain uncharacterized
ORFs and may suffer from subsequent additions, deletions, and modifications.
In general, incompleteness of a cellular subsystem defined by a set of
genes or gene products taken from the complete catalog is quite suggestive
for further investigation, as in those missing enzymes in a specific metabolic
pathway. The concept of the minimal gene set is another example of the
power of cross-species comparison of complete catalogs (Mushegian &
Koonin, 1996). As the computational analyses play major roles in functional
genomics, the management of the functional, albeit predicted, data also
requires a major investment because of frequent updates. In addition to
the PATHWAY database, KEGG maintains the GENES database that is a collection
of gene catalogs for many organisms. Each gene catalog is hierarchically
classified according to the most up-to-date functional assignment and all
the catalogs are correlated by the orthologous gene table. With the development
of the KEGG databases, the computational techniques such as for path computation
and network comparison will become increasingly useful in automating reasoning
steps that mimic biologists*f logics in silico.
Acknowledgment
This work was supported in part by
a Grant-in-Aid for Scientific Research on Priority Areas, *eGenome Science*f,
from the Ministry of Education, Science, Sports and Culture of Japan. The
computation time was provided by the Supercomputer Laboratory, Institute
for Chemical Research, Kyoto University.
References
Brenner, S.E., Hubbard, T., Murzin, A., and Chothia, C., 1995, Gene duplication in H. influenzae, Nature 378, 140.
Brutlag, D.L., Galper, A.R., and Millis, D.H., 1991, Knowledge-based simulation of DNA metabolism: prediction of enzyme actions. Comput. Appl. Biosci. 7, 9-19.
Cohen, D.M., and Bergman, R.N., 1994, SYNTAX: a rule-based stochastic simulation of the time-varying concentrations of positional isotopomers of metabolic intermediates. Comput. Biomed. Res. 27, 130-147.
Danchin, A., 1997, Comparison between the Escherichia coli and Bacillus subtilis genomes suggests that a major function of polynucleotide phosphorylase is to synthesize CDP. DNA Res. 28, 9-18.
Franco, R. and Canela, E.I., 1984, Computer simulation of purine metabolism. Eur. J. Biochem. 144, 305-315.
Fujibuchi, W., Goto, S., Migimatsu, H., Uchiyama, I., Ogiwara, A., Akiyama, Y., and Kanehisa, M., 1997, DBGET/LinkDB: an Integrated Database Retrieval System. Pacific Symposium on Biocomputing *f98, in press.
Gaasterland, T., Godfrey, P., and Minker, J., 1992, An overview of cooperative answering. J. Intelligent Information Systems 1, 123-157.
Gerhard, M. (ed.), 1992, Biological Pathways, Third Edition (Boehringer Mannheim).
Goto, S., Bono, H., Ogata, H., Fujibuchi, W., Nishioka, T., and Kanehisa, M., 1996, Organizing and computing metabolic pathway data in terms of binary relations. Pacific Symposium on Biocomputing *f97, pp. 175-186.
Hofest_dt, R., and Meineke, F., 1995, Interactive modelling and simulation of biochemical networks. Comput. Biol. Med. 25, 321-334.
Hubbard, T.J.P., New horizons in sequence analysis, 1996, Curr. Opin. Struct. Biol. 7, 190-193.
Kanehisa, M., 1997a, A database for post-genome analysis. Trends Genet. 13, 375-376.
Kanehisa, M., 1997b, Linking databases and organisms: GenomeNet resources in Japan. Trens Biochem. Sci., in press.
Karp, P.D., Riley, M., Paley, S.M., and Pelligrini-Toole, A., 1996, EcoCyc: an encyclopedia of Esherichia coli genes and metabolism. Nucleic Acids Res. 24, 32-39.
Koonin, E.V., 1997, Big time for small genomes. Genome Res. 7, 418-421.
Luban, J., and Goff, S.P., 1995, The yeast two-hybrid system for studying protein-protein interactions, Curr. Opin. Biotechnol. 6, 59-64.
Mavrovouniotis, M.L., 1995, Computational methods for complex metabolic systems: Representation of multiple levels of detail, in: Bioinformatics & Genome Research, Lim, H.A. and Cantor C.R. (eds.) (World Scientific) pp. 265-273
Mushegian, A.R., and Koonin, E.V., 1996, A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc. Natl. Acad. USA 93, 10268-10273.
Nishizuka, T. (ed.), 1980, Metabolic Maps (Biochemical Society of Japan).
Nishizuka, T. (ed.), 1997, Cell Functions and Metabolic Maps (Biochemical Society of Japan).
Okamoto, M., Morita, Y., Tominaga, D., Tanaka, K., Kinoshita, N., Ueno, J.-I., and Miura, Y., 1996, Toward a virtual-labo-system for metabolic engineering: Development of biochemical engineering system analyzing tool-kit (BEST-KIT). Pacific Symposium on Biocomputing *f97, 304-315.
Ogata, H., Bono, H., Fujibuchi, W., Goto, S., and Kanehisa, M., 1996, Analysis of binary relations and hierarchies of enzymes in the metabolic pathways. Proc. 7th Workshop on Genome Informatics, 128-136.
Otsuka, J., Watanabe, H., and Mori, K.T, 1996, Evolution of transcriptional regulation system through promiscuous coupling of regulatory proteins with operons; suggestion from protein sequence similarities in Escherichia coli. J. theor. Biol. 178, 183-204.
Selkov, E., Basmanova, S., Gaasterland, T., Goryanin, I., Gretchkin, Y., Maltsev, N., Nenashev, V., Overbeek, R., Panyushkina, E., Pronevitch, L., Selkov, E., Jr., and Yunus, I., 1996, The metabolic pathway collection from EMP: the enzymes and metabolic pathways database. Nucleic Acids Res. 24, 26-28.
Schomburg, D. and Salzmann, M. (eds.), 1990 Enzyme Handbook (Springer-Verlag).
Suyama, M., Ogiwara, A., Nishioka, T., and Oda, J., 1993, Searching for amino acid sequence motifs among enzymes: the Enzyme-Reaction Database. Comput. Appl. Biosci. 9, 9-15.
Tamames, J., Casari, G., Ouzounis, C., Valencia, A., 1997, Conseverd clusters of functionally related genes in two bacterial genomes. J. Mol. Evol. 44, 66-73.
Woese, C.R., Kandler, O., and Wheelis, M.L., 1990, Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 87, 4576-4579.
Wolf, K., and Butler, G., 1997, Yeast*fs
clickable chromosomes and other genome browsers. Trends Genet. 13, 246-247.